
«У нас не было никаких подозрений, что ребёнок какой-то не такой»
Целый год до постановки диагноза Астемир ходил в обычный детский сад. Правда, с первых же дней заведующая предупредила маму мальчика: «Дети у нас очень подвижные, мы беспокоимся за Астемира. Его могут нечаянно толкнуть и уронить. Не хотите ли вы перевести его в специальную группу?».
![]()
Летом 2022 года у Астемира был выпускной в детском саду.
Астемир и правда был в два раза меньше остальных, чем очень нравился девочкам, которые называли его «наш сыночек». Предложение о спецгруппе смутило Любовь: если ребенок умственно развивается по нормам, зачем ограничивать его общение? И Астемир продолжил ходить в обычный садик.
Когда же поставили диагноз «прогерия», врачи Сеченовского Университета посоветовали поберечь ребенка, так как даже обычная простуда может дать резкий старт другому заболеванию. Тогда Любовь забрала сына из детского сада. С тех пор Астемир ходил туда только на утренники и другие детские праздники
Как любому другому ребенку, ему нравится внимание, игры и общение.
«Астемиру уже семь, но в школу он пойдет только на следующий год вместе с младшим братом. Я не рискую их разделять. — рассказывает Любовь. — Мы с мужем относимся к Астемиру как к обычному здоровому ребенку, не делаем акцент на его особенностях. У детей с прогерией жизнь очень быстро проходит, поэтому просто нужно ловить каждый момент, находясь рядом, и радоваться тому, что у тебя есть».
Лечение синдрома Марфана
К сожалению, на сегодняшний день лекарственные методы терапии этой генетической патологии еще не разработаны
Однако пациентам с синдромом Марфана важно соблюдать все назначения врачей, чтобы устранить симптомы патологии и замедлить темпы ее развития
Лечение зависит от клинических проявлений болезни:
- при аневризме аорты назначают препараты, которые снижают частоту и силу сердечных сокращений, снимая избыточную нагрузку на сосуды;
- пациентам с синдромом Марфана часто назначают антигипертензивные препараты для снижения артериального давления;
- хондроитин и глюкозамин относятся к естественным компонентам соединительной ткани — их прием улучшает структуру хрящей и предупреждает патологии суставов;
- для стимуляции образования коллагена выписывают специальные БАДы — L-карнитин, витамины из групп С, D, Е, В, а также кальций, цинк и другие пищевые добавки.
Пациентам противопоказаны физические нагрузки, постоянная активность, травмоопасные игры. Рацион питания людей с синдромом Марфана должен быть насыщен белками, полезными жирными кислотами, микро- и макроэлементами. Для поддержки структур скелета пациентам с мутацией в гене FBN1 показано ношение корсетов, укрепление мышц с помощью ЛФК и оздоровительного массажа.
В некоторых случаях может помочь только хирургическое лечение — операции по замене части аорты, клапанов, исправлению костных патологий или коррекции патологий глаза, которые существенно снижают риски опасных осложнений.
Лента новостей
c http-equiv=»Content-Type» content=»text/html;charset=UTF-8″>lass=»infinite-scroll-component__outerdiv»>
Все новости
Екатерина Неженцева: «Я счастливый человек, но от «пластики» не отказалась бы»
![]()
В семье Екатерины Неженцевой растут два сына-помощника, во всем поддерживают и помогают маме. Фото: из архива Екатерины Неженцевой
Лет пять назад о Екатерине Неженцевой из поселка Уренгой писали многие федеральные СМИ, ее приглашали на ток-шоу, брали интервью. Потом шумиха утихла. Как сегодня живет Екатерина, что изменилось в ее судьбе, узнал «Красный Север».
Время для чтения ~ 6 минут
Доброе детство Екатерины Неженцевой и диагноз «прогерия»
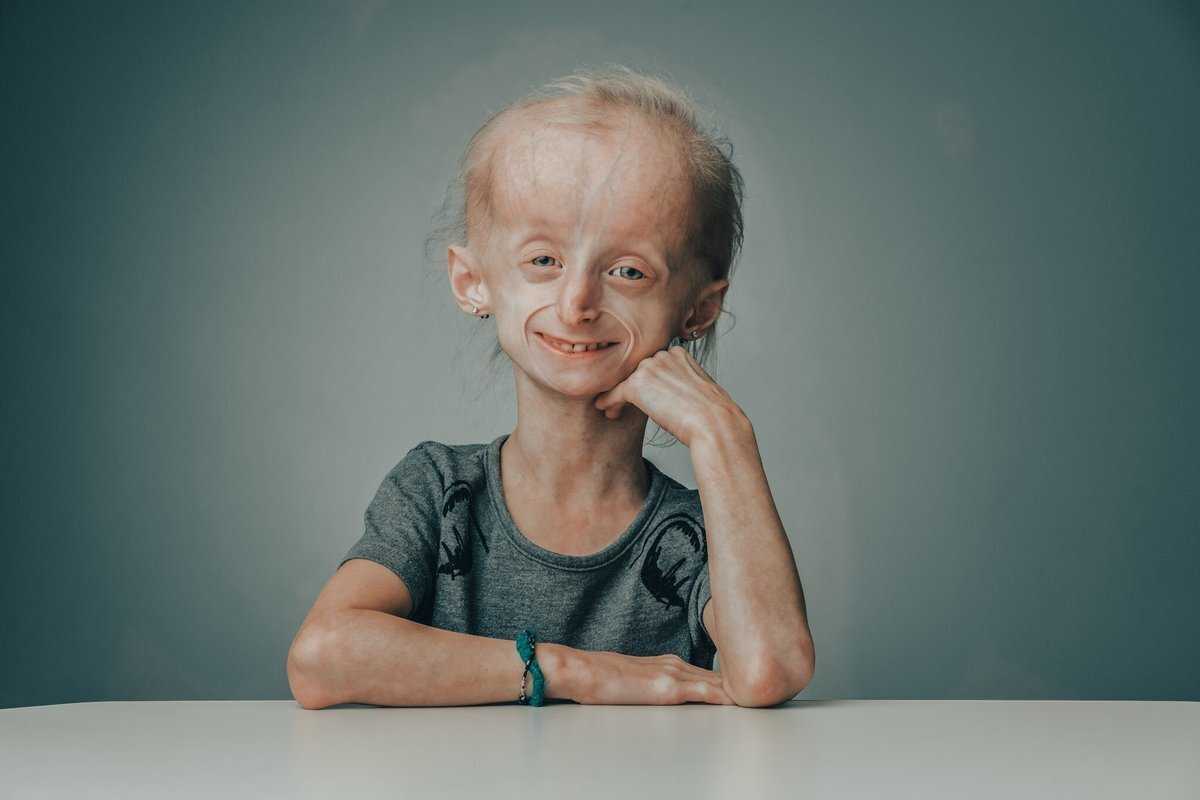
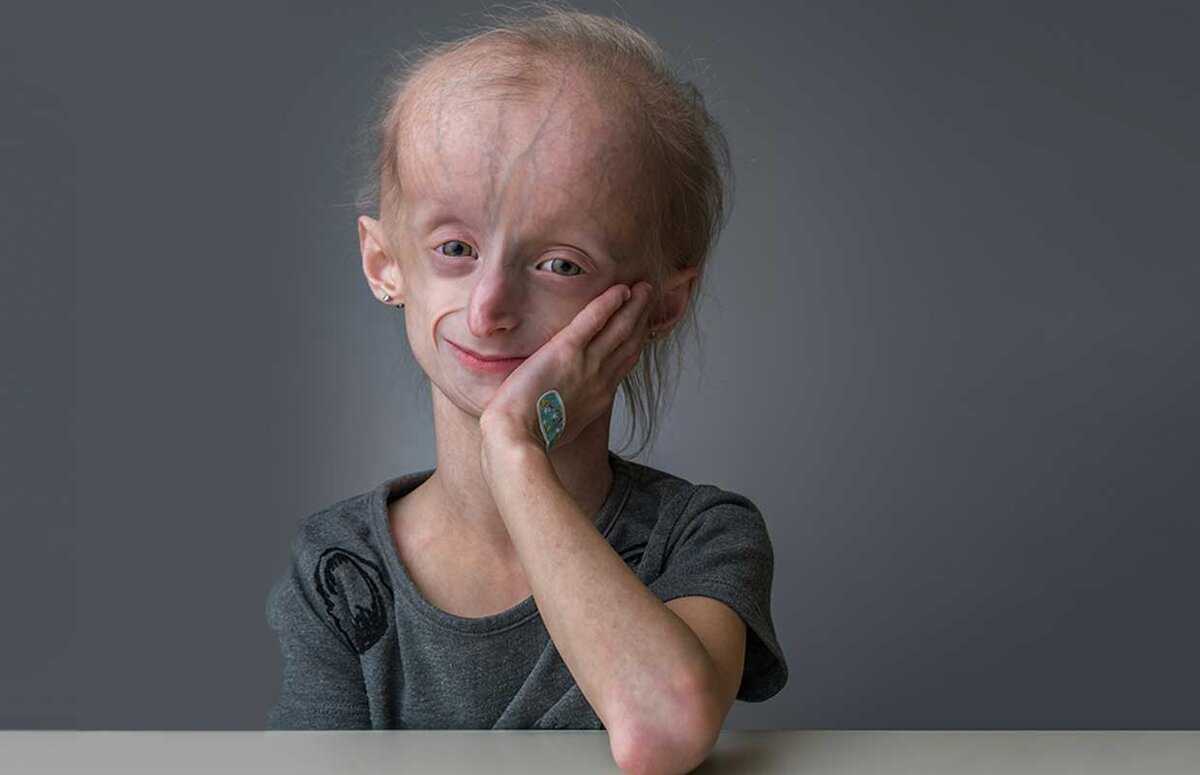
У Кати, уроженки Лимбяяхи (отдаленный район Нового Уренгоя. — Прим. ред.), генетическое заболевание. Когда девочка родилась, близкие сразу заподозрили неладное. Первой тревогу забила бабушка, заметив у малышки морщины. С каждым месяцем их становилось больше, и родители пошли по врачам. Обследования ничего не дали, кроме неутешительного диагноза «прогерия». Лечения нет, лишь поддерживающая терапия. С такой болезнью люди долго не живут, от силы до 13—14 лет, так как организм стремительно стареет…
Лишь годы спустя, Катя и ее семья смогли выдохнуть спокойно — прогнозы докторов не подтвердились.
![]()
Первоначальный диагноз «прогерия» не подтвердился. Фото: из архива Екатерины Неженцевой
Отец бросил семью сразу же, как узнал о диагнозе дочери. Где он и что с ним, Катя не знает до сих пор. Мама вышла во второй раз замуж, родила Екатерине братика и сестричку. В детском саду и школе Катя не испытывала особых проблем. Общительная и доброжелательная девочка легко сходилась с людьми. Она хорошо училась, и с теплотой вспоминает детские годы.
— Наверное, мой характер сформировали мама и бабушка. Они меня сделали настоящим бойцом, научили ценить то, что есть, и всегда верили, что я буду счастливой, — рассказала Екатерина «Красному Северу».
Ток-шоу и пустословные обещания. Кто помог Екатерине Неженцевой
Мама всеми силами старалась помочь дочери. Поэтому первый раз Катя попала на ток-шоу, когда ей было 10 лет. На программе «Малахов+» эксперты обещали, что сделают школьнице операцию, но это оказалось пустословием. В 16 лет Катя стала героиней ток-шоу «Пусть говорят», и снова хирурги давали обещания и ничего не сделали, только выставили космический счет, который при всем желании ямальская семья не смогла бы оплатить.
Но мир не без добрых людей. Нашлись благотворитель, который помог деньгами, и медики из Омска, которые выполнили несколько операций за божескую цену.
Девушка преобразилась. К тому времени она училась в техникуме, но не успела его закончить, вышла замуж, родила сынишку Глеба, следом еще одного — Михаила. К сожалению, у младшего оказалась та же болезнь, что и у мамы. С мужем не сложилось.




![]()
Михаил, сынишка Екатерины, унаследовал болезнь мамы, но не унывает, растет любознательным и веселым. Фото: из архива Екатерины Неженцевой
На тот момент Катя уже знала, что прогнозы врачей неверны. На медицинском языке ее болезнь называется синдромом вялой кожи — таких людей в мире единицы. Недуг не затронул ее тело, только лицо, а все органы соответствуют биологическому возрасту. Жить она будет долго и, уверена девушка, счастливо.
Екатерина Неженцева: «Счастье — не подарок судьбы, а выбор человека»
Сейчас Кате 28 лет, она не любит общаться с журналистами. Не хочет выставлять свою жизнь напоказ, редко рассказывает о личном, только говорит, что она не одинока. Впрочем, это неудивительно: у девушки никогда не было недостатка в поклонниках.
В Новом Уренгое Екатерина — знаменитость, ее узнают, иногда просят вместе сфотографироваться, относятся доброжелательно. Старший сын помогает во всем, растет заботливым и внимательным. Младшему 7 лет, он учится в той же школе, что когда-то и мама.
— У него есть друзья, к нему относятся, как к любому другому мальчишке в классе, — рассказывает Екатерина. — Миша еще мал, поэтому я не думаю об операции для него.
![]()
Цветы, которые делает Екатерина, радуют своей яркостью. Фото: из архива Екатерины Неженцевой
Екатерина работает делопроизводителем в нефтяной компании, ездит в Коротчаево (отдаленный район Нового Уренгоя. — Прим.ред.). Есть у нее хобби — делать цветы из фоамирана.
— На ток-шоу я встретилась с разными людьми, некоторые глубоко несчастные, без надежды в глазах. Я думаю, что счастье, хорошее настроение — не подарок судьбы, а выбор самого человека. У меня все есть: дети, мама, отчим, любимый человек, работа. Конечно, было бы неплохо, если бы нам дали более просторную квартиру — я стою в очереди, как инвалид детства, на расширение жилплощади. Не отказалась бы от пластической операции, если найдется хирург, который возьмется за мое лицо.
Старость начинается в детстве
«Мы приехали к врачу в Ставрополь, и как только мы переступили порог кабинета, он сразу сказал: „У вас прогерия!“ И достал большую медицинскую энциклопедию, чтобы показать нам это заболевание», — рассказывает мама Астемира Любовь. Со страниц книги на родителей мальчика смотрели дети, очень похожие на их сына. Что было дальше, женщина не помнит, ее охватил ужас. Все четыре часа пути домой в Нальчик в глазах стояли слезы.
Для подтверждения диагноза в декабре 2020 года у Астемира взяли кровь на анализ в медико-генетическом центре Ставрополя, образец отправили в Москву. Все новогодние праздники Любовь с мужем не говорили о болезни сына, каждый сам по себе искал информацию в интернете и всей душой надеялся, что все обойдется. «В какой-то момент мы сказали друг другу: у нас это не подтвердится. И выдохнули», — вспоминает Любовь.
![]()
Астемир с мамой.
Результат родителям ребенка сообщили 12 января 2021 года. У Астемира официально диагностировали страшную болезнь прогерию или синдром Хатчинсона-Гилфорда — очень редкое генетическое заболевание, которое встречается у одного человека из 20 миллионов.
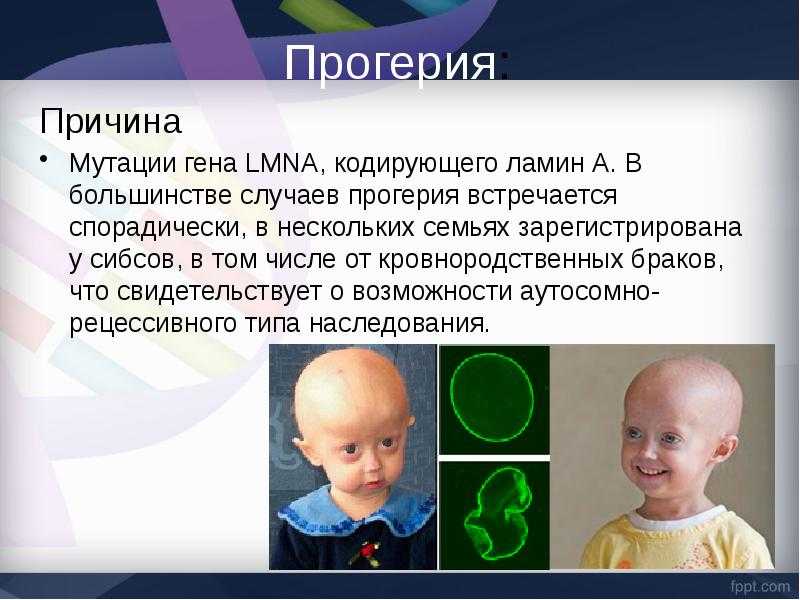
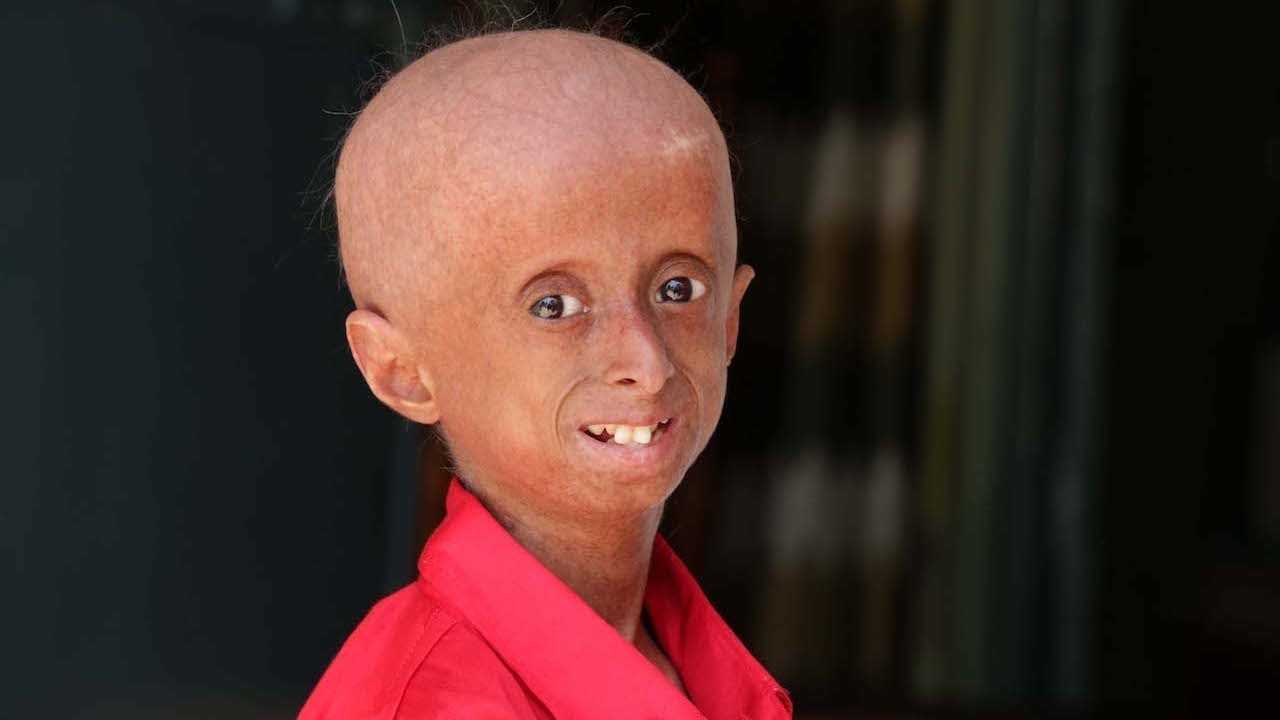
Причиной этой болезни является мутация гена LMNA, в результате которой вырабатывается токсичный белок прогерин, вызывающий быстрое старение и сокращающий продолжительность жизни. Прогерию редко диагностируют при рождении, так как основные симптомы болезни проявляются после двух лет. Изменяется внешность, состояние кожи, развиваются тяжелые осложнения, в особенности атеросклероз и его последствия: инфаркт миокарда, инсульт, сердечная недостаточность. Продолжительность жизни у детей с прогерией обычно составляет до подросткового возраста или до двадцати лет.
Чтобы подтвердить диагноз «прогерия», врачи-генетики проверяют определенные участки гена LMNA (так называемые «горячие точки»), изменения в которых приводят к продукции токсичного прогерина.
«Также существует группа очень схожих заболеваний, которые называются прогероидным синдромом или прогероидными ламинопатиями, — говорит Юлия Тихонович, кандидат медицинских наук, заведующая отделением детской эндокринологии Центра Материнства и Детства ФГАОУ ВО Первый МГМУ им. И.М. Сеченова МЗ РФ. — Они отличаются от прогерии тем, что при них не вырабатывается прогерин. Эти заболевания могут протекать тяжелее или легче, чем классическая прогерия, а встречаться еще реже. Когда необходимо подтвердить или исключить „прогероидный синдром“, проводится полное секвенирование гена LMNA или других генов».
Прогерия входит в перечень орфанных заболеваний, который Министерство здравоохранения России формирует и ежегодно обновляет на основе статистики. В 2023 году перечень включает 273 пункта, среди которых есть как отдельные заболевания, так и целые группы.
Причины возникновения прогерии
![]()
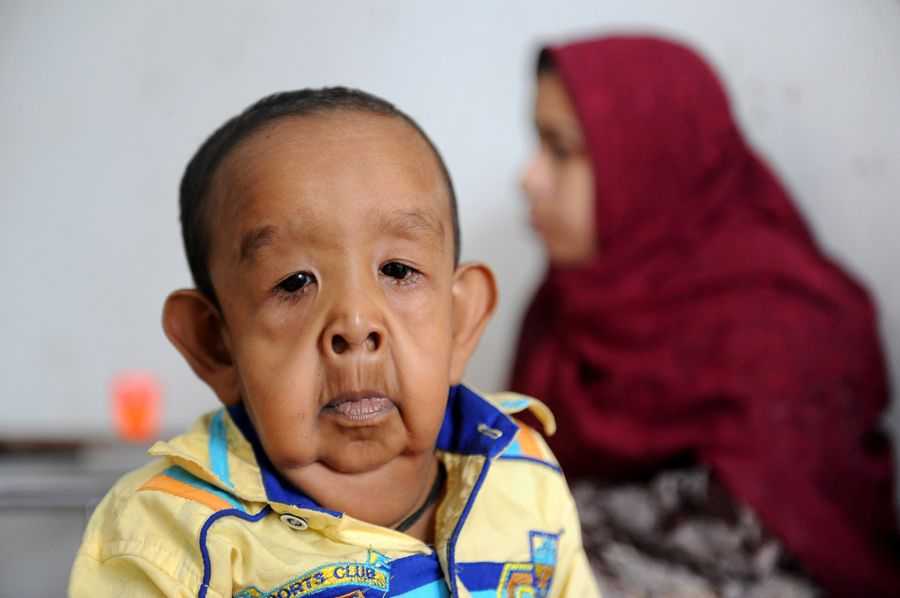
Сегодня науке удалось выявить причину такого заболевания. У детей прогерия появляется из-за мутации гена LMNA, отвечающего за кодировку белков-ламинов. В некоторых семьях прогерия встречается у детей одних и тех же родителей (например, Анджали Кумари и Кешав Кумари, видео в конце статьи), иногда ими являются кровные родственники, что тоже может являться причиной сбоя в наследственности.
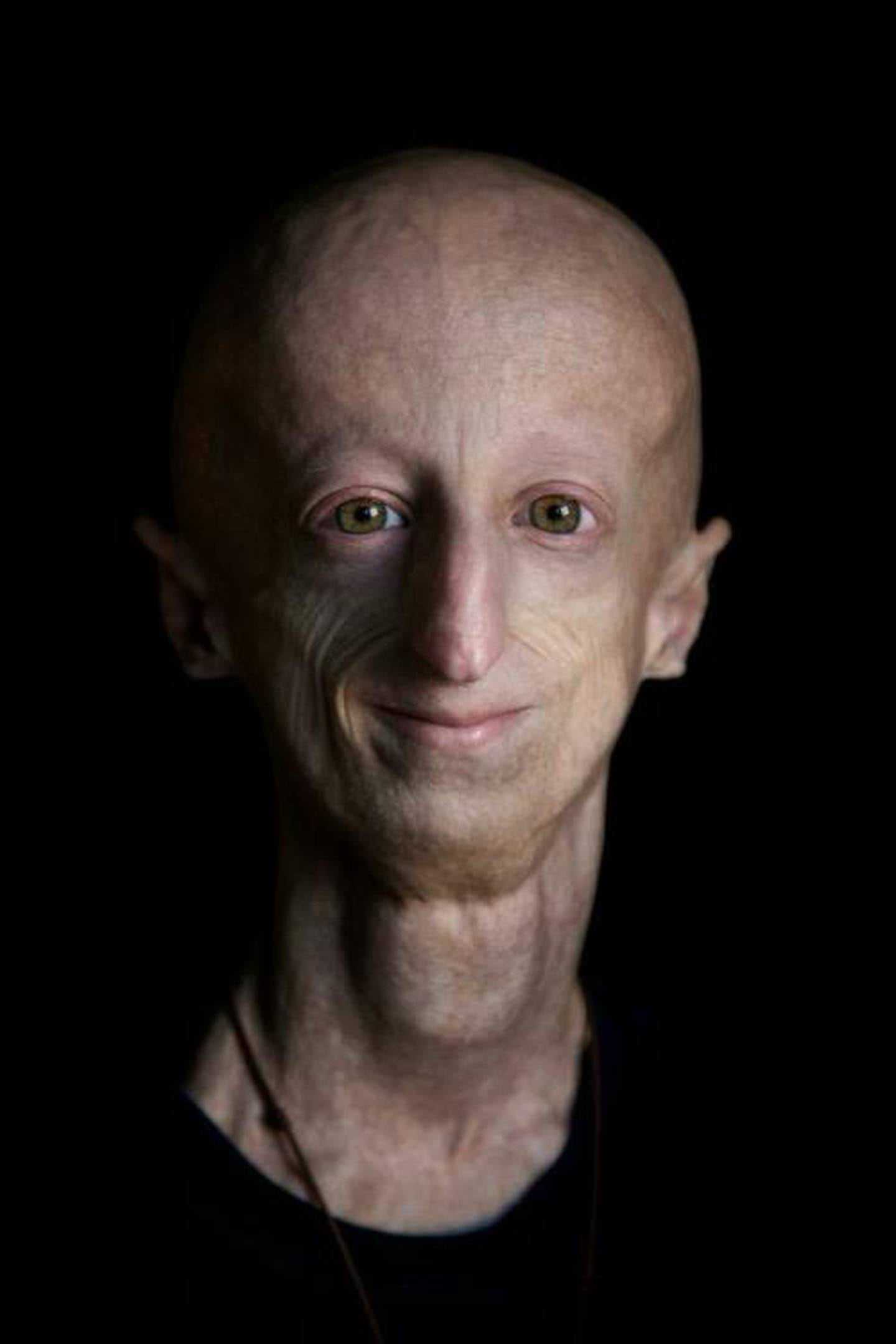
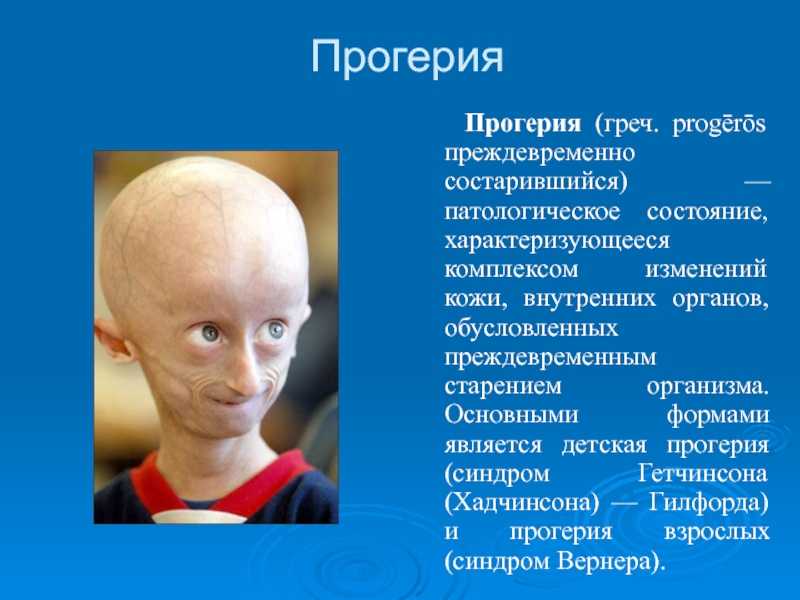
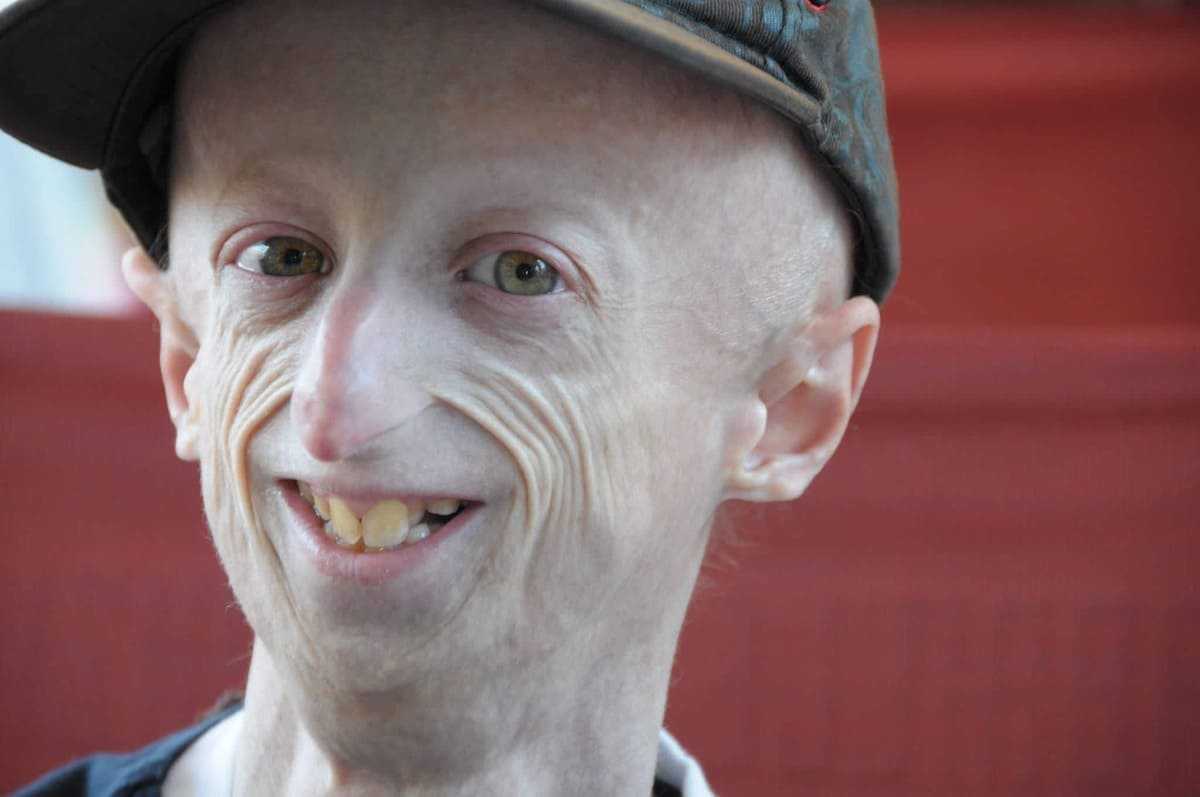
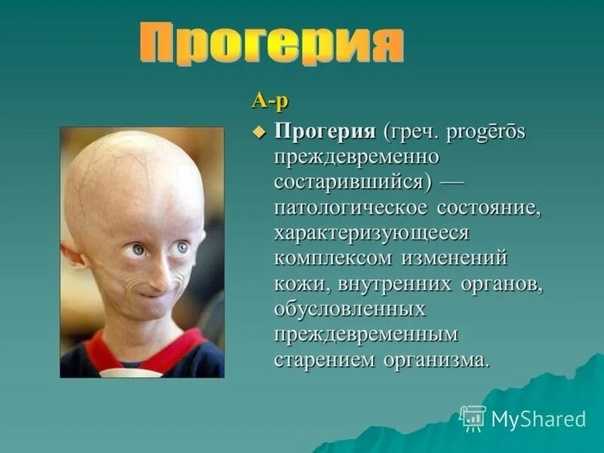
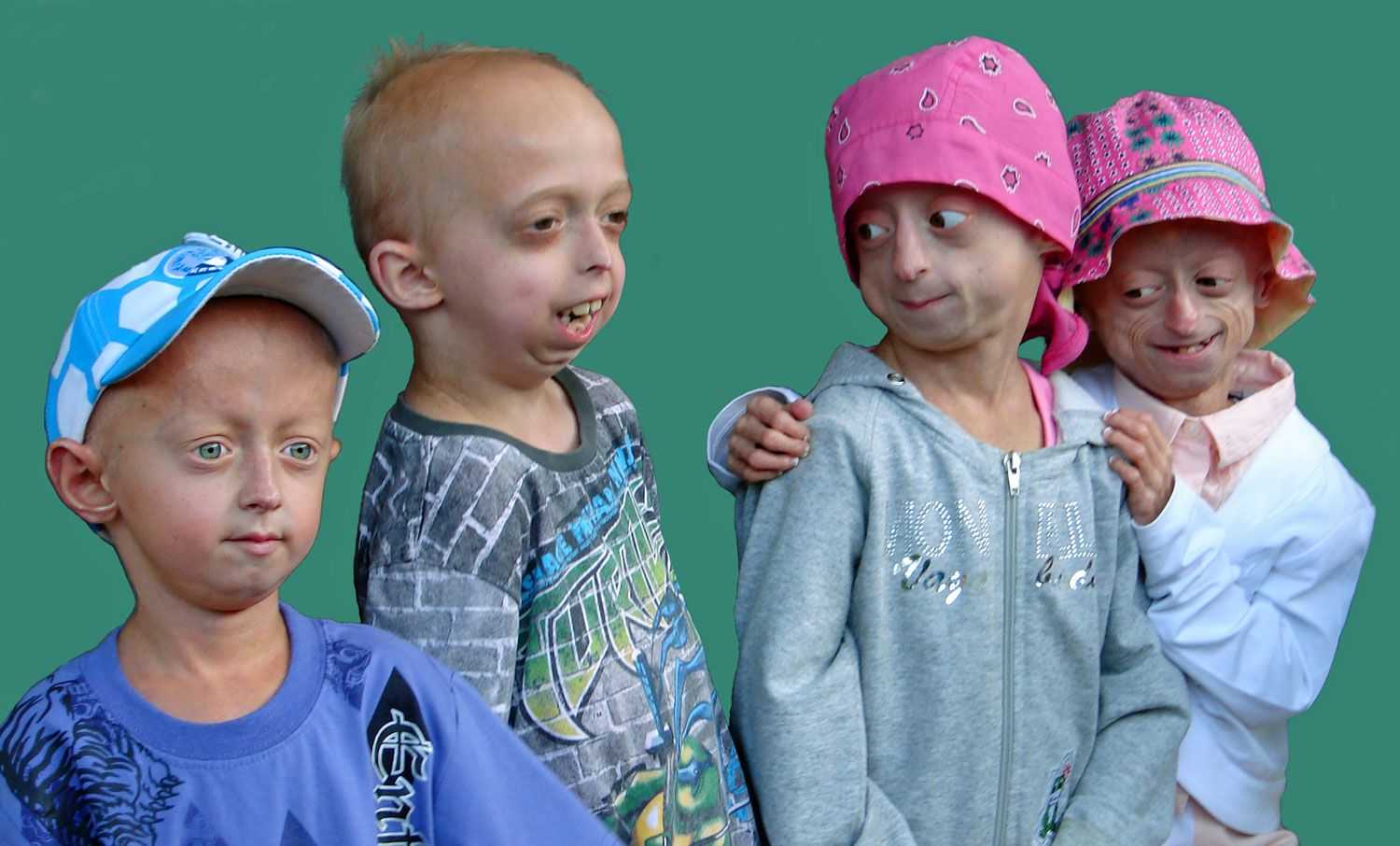
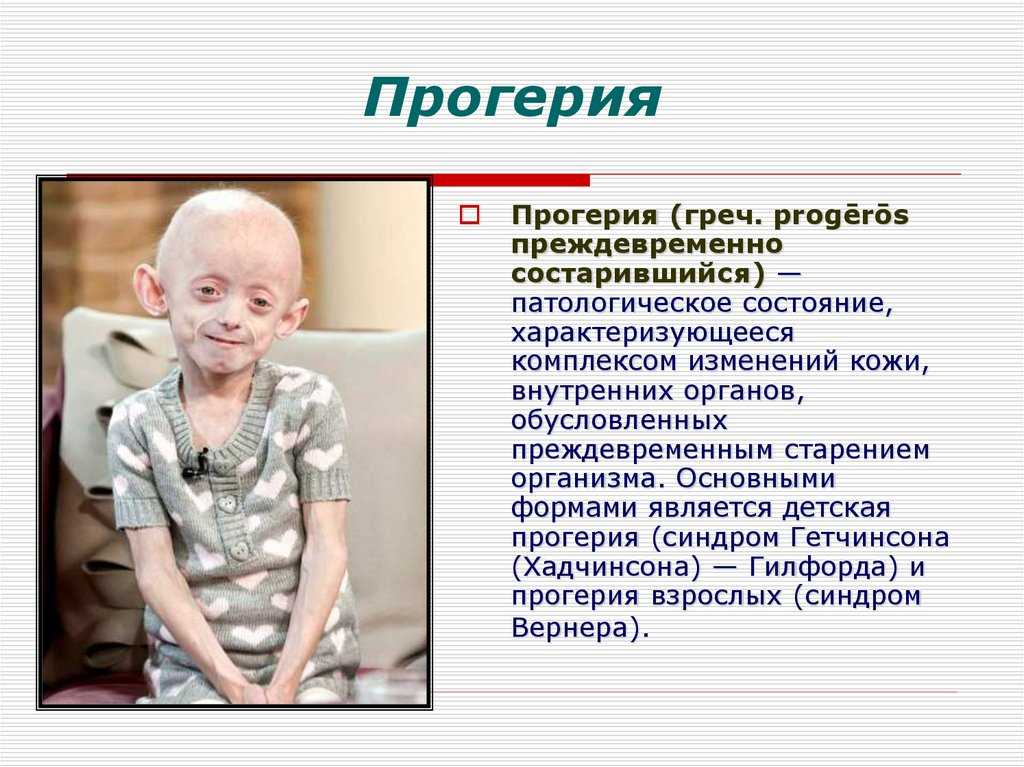
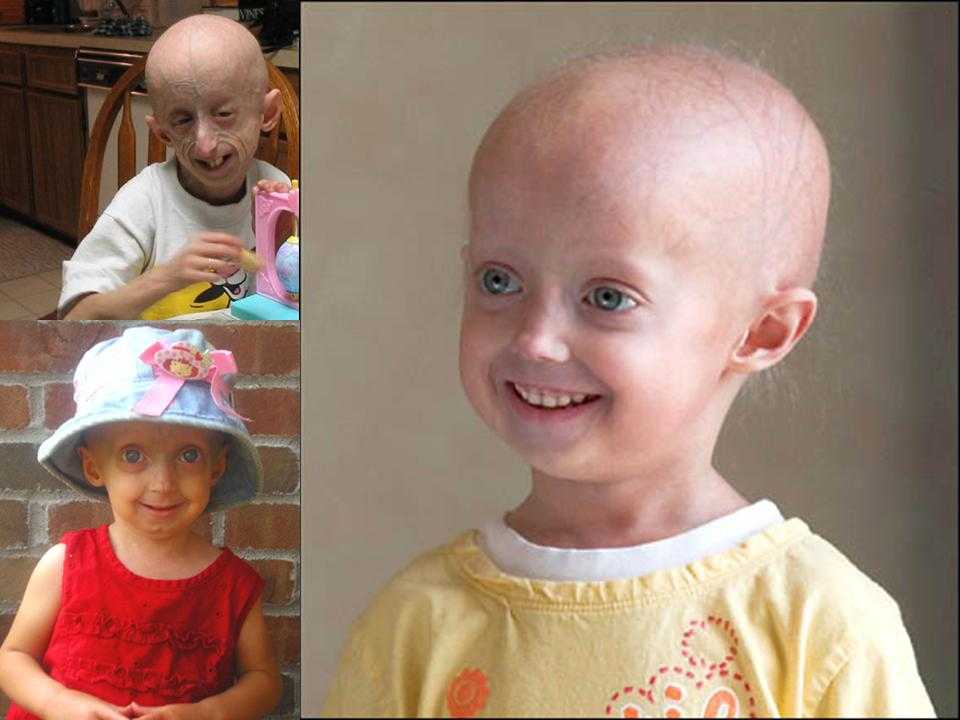
Прогерия чаще всего не проявляется с рождения, ее признаки дают о себе знать на 2-3 году жизни. Ребенок снижает темпы роста, резко меняется состояние кожи и подкожной клетчатки. Поверхность тела становится сухой и морщинистой, начинают просвечиваться вены. У ребенка увеличивается голова, но не лицо, а нижняя челюсть остается недоразвитой. Развитие организма тоже происходит с явными нарушениями: атрофируются мышцы, выпадают зубы, волосы и ногти, слабым становится костный аппарат и суставы, прекращаются развиваться половые органы, ухудшается зрение.
К сожалению, с таким набором болезней человеку трудно бороться – средняя продолжительность жизни при детской прогерии составляет всего 13 лет. Смерть наступает в возрасте от 7 до 27 лет, в истории медицины известны только 2 пациента, прожившие дольше 27 лет.
![]()
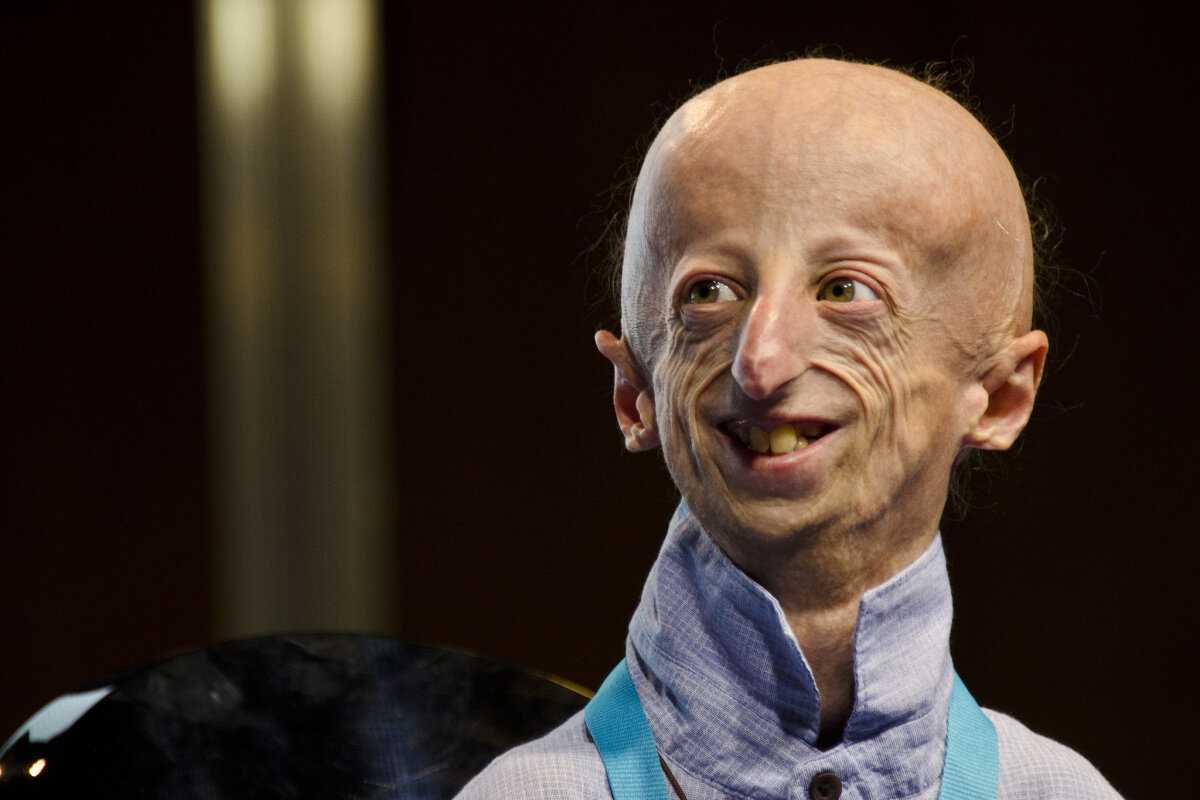
Прогерия у взрослых обусловлена нарушением гена WRN. Проявляется заболевание в период полового созревания человека, что приводит к замедлению его роста. На третьем десятке жизни начинают седеть и выпадать волосы, развивается катаракта, тонкой становится кожа. На ней появляются язвы. Ротовое отверстие и нос становятся узкими, тем самым лицо начинает быть похожим на маску. В 30-40 лет проявляет себя сахарный диабет, атеросклероз, возможны и злокачественные опухоли. К сожалению, общего лечения нет – врачи борются с конкретными осложнениями в зависимости от силы симптомов. И в данном случае прогерия заканчивается смертью из-за многочисленных осложнений сосудистой системы и злокачественных образований.
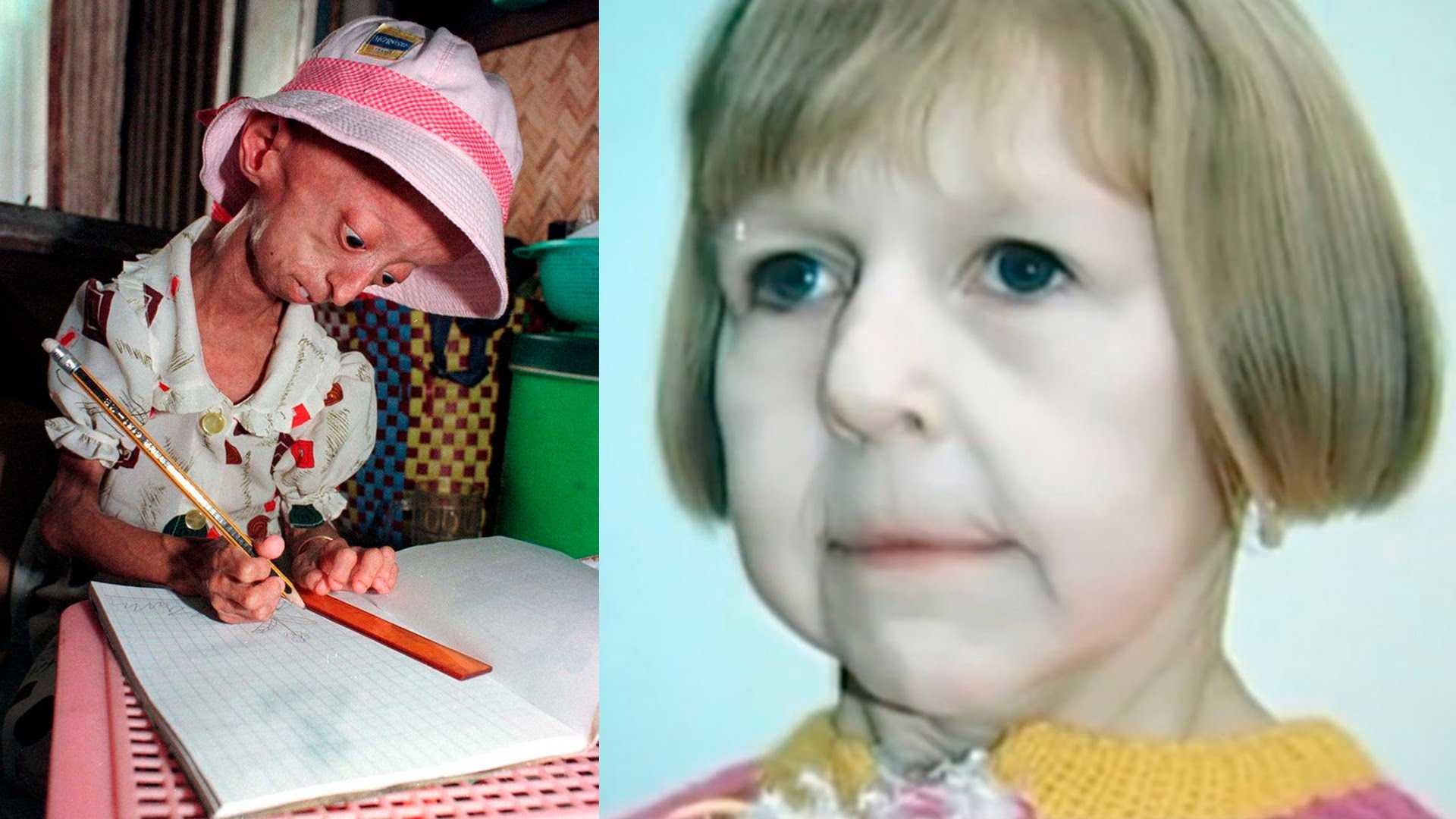
Уже установлено, что прогерия напрямую связана с молекулярными изменениями в организме, происходящими во время старения. Заболевание называют «синдромом Бенджамина Баттона» в честь киноперсонажа, тоже выглядевшим пожилым в детском возрасте.
К сожалению, человечество еще не нашло способа бороться со старостью, да и нужно ли это? Увы, но некоторые люди начинают стареть не душой, а телом еще в детстве. Но история Сэма Бернса показывает, что любовь к жизни все равно сильнее страха смерти. Американский подросток, который умер от прогерии в возрасте всего 17 лет, много выступал с мотивационными лекциями. Сэм публично рассказывал, как даже с физическими ограничениями пытается радоваться каждому прожитому дню и осуществлять свои мечты.
Что такое «прогерия»?
Для начала расшифруем само слово «прогерия». Оно составлено из двух греческих слов – «сверх» и «старик». Таким образом больные становятся «сверхстариками», что выглядит удручающе. Прогерия может быть, как детской, так и взрослой. Впервые такой случай был описан еще в 1886 году английским врачом Хатчинсоном. Он наблюдал за шестилетним мальчиком, который начал быстро стареть, что сопровождалось отмиранием кожи. На основе этой истории и появился в медицине термин «прогерия». Аналогичное заболевание у взрослых было описано чуть позже – немецкий врач Карл Вернер на основе наблюдений за четырьмя пациентами защитил докторскую диссертацию в 1904 году. Прогерия – редчайшая болезнь, всего зафиксировано около 350 ее случаев.
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.

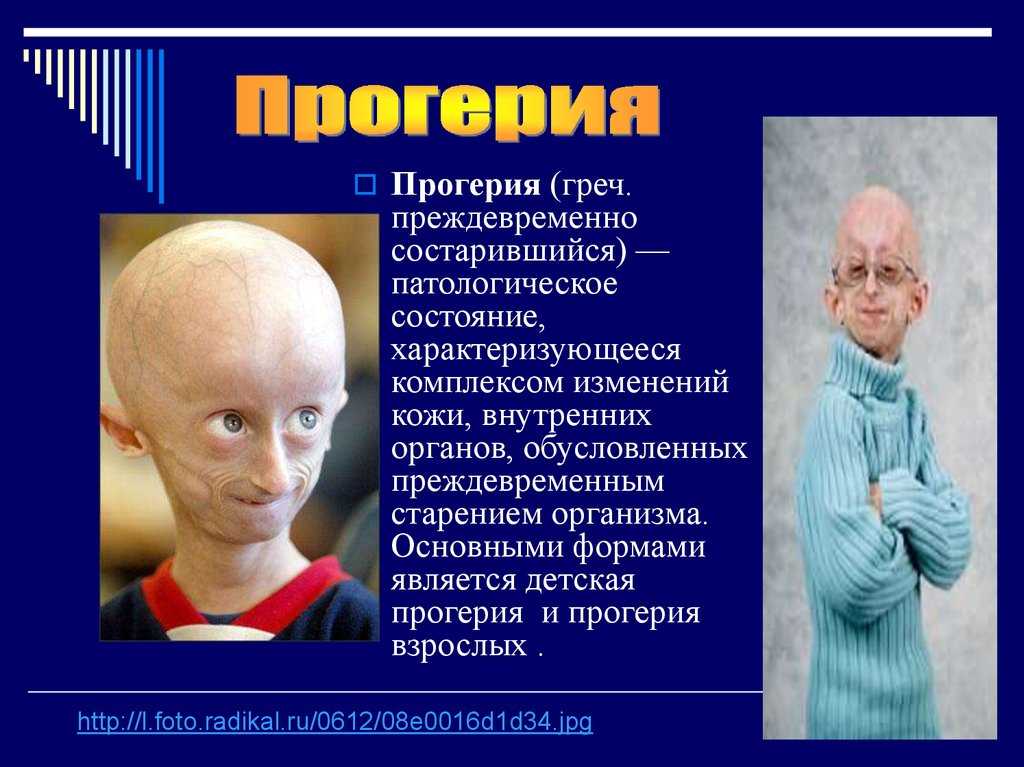




Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на .
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно
Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.
Классификация синдрома Марфана
Выделяют несколько форм заболевания в зависимости от особенностей клинических проявлений генетической мутации.
Существуют две основные клинические формы патологии:
- Стертая. Таким пациентам «везет» больше: аномалия у них проявляется поражениями только одной-двух систем организма, а симптомы выражены незначительно. Люди могут жить практически нормальной жизнью, несмотря на болезнь.
- Выраженная. В таких случаях поражаются три и более систем организма, либо значительно нарушается функционирование одной из систем.
В зависимости от степени проявления выделяют легкие, среднетяжелые и тяжелые формы синдрома Марфана. Тяжелые патологии встречаются гораздо реже: частота их выявления составляет примерно 1 на 25–50 тысяч человек.
Принципиальную роль в определении прогноза болезни играет характер ее течения:
- Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, а с каждым годом жизни пациента возрастают риски фатальных осложнений.
- Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Выделяют три разных, но похожих заболевания:
- Синдром Марфана — стертая форма патологии с положительным результатом генетического тестирования.
- Болезнь Марфана — классическая клиническая картина с подтвержденным семейным наследованием.
- Марфаноподобный синдром — проявление патологии соединительной ткани без генетической мутации.
Первые признаки заболевания чаще всего проявляются еще в детском возрасте. К подростковому периоду становится понятно, насколько быстро у пациента прогрессирует болезнь, вызванная мутацией гена FBN1.
Екатерина Неженцева: «Счастье — не подарок судьбы, а выбор человека»
yle=»text-align:» class=»Typography_text__WDByQ Typography_size__15__DJDOw Typography_none__FajqV Typography_primary__29LdH «>Сейчас Кате 28 лет, она не любит общаться с журналистами. Не хочет выставлять свою жизнь напоказ, редко рассказывает о личном, только говорит, что она не одинока. Впрочем, это неудивительно: у девушки никогда не было недостатка в поклонниках.
В Новом Уренгое Екатерина — знаменитость, ее узнают, иногда просят вместе сфотографироваться, относятся доброжелательно. Старший сын помогает во всем, растет заботливым и внимательным. Младшему 7 лет, он учится в той же школе, что когда-то и мама.
— У него есть друзья, к нему относятся, как к любому другому мальчишке в классе, — рассказывает Екатерина. — Миша еще мал, поэтому я не думаю об операции для него.
![]()
Цветы, которые делает Екатерина, радуют своей яркостью. Фото: из архива Екатерины Неженцевой
Екатерина работает делопроизводителем в нефтяной компании, ездит в Коротчаево (отдаленный район Нового Уренгоя. — Прим.ред.). Есть у нее хобби — делать цветы из фоамирана.
— На ток-шоу я встретилась с разными людьми, некоторые глубоко несчастные, без надежды в глазах. Я думаю, что счастье, хорошее настроение — не подарок судьбы, а выбор самого человека. У меня все есть: дети, мама, отчим, любимый человек, работа. Конечно, было бы неплохо, если бы нам дали более просторную квартиру — я стою в очереди, как инвалид детства, на расширение жилплощади. Не отказалась бы от пластической операции, если найдется хирург, который возьмется за мое лицо.
Следить за сосудами и беречь сердце
Детей с прогерией наблюдают в российских федеральных центрах. Один из таких — Клинический центр Первого Московского государственного медицинского университета имени И.М. Сеченова (Сеченовский Университет). Именно туда на обследование каждые три-четыре месяца ездит Астемир вместе с мамой.
![]()
Астемиру нравится летать в Москву на самолёте и гулять по большому городу.
Полное обследование организма необходимо, чтобы вовремя заметить признаки какой-либо сопутствующего осложнения и начать симптоматическое лечение. К счастью, у Астемира пока нет серьезных проблем со здоровьем, кроме генетического диагноза.
Прогерия же опасна тем, что быстро изнашивает сердце и сосуды, отчего даже у маленьких детей случаются инфаркты и инсульты. Для их профилактики детям необходимо ограничить физические нагрузки и принимать специальный лекарственный препарат с действующим веществом лонафарнибом. Это лекарство было создано в США специально для детей с прогерией. Его разработку инициировали супруги Лесли Гордон и Скотт Бернс, основавшие в 1999 году исследовательский фонд Progeria.
Лонафарниб помогает блокировать накопление прогерина и препятствует повреждению клеток и развитию проблем с сердцем и костями. В клинических исследованиях препарата приняли участие больше 90 детей и подростков. По прошествии 11 лет исследования ученые сделали вывод, что лонафарниб позволяет продлить жизнь в среднем на 2,5 года.
Пока что это единственное доступное лекарство для людей с прогерией. Однако сегодня также ведутся разработки генной терапии. В январе 2021 года в журнале Nature были опубликованы предварительные результаты исследования на мышах нового генотерапевтического препарата против прогерии. Учёные создали его на основе аденоассоциированного вируса, способного доставить в организм здоровую копию дефектного гена.
Одна инъекция этого препарата существенно снизила уровень прогерина, повысила жизнеспособность и увеличила продолжительность жизни мышей с 215 до 510 дней. Эти результаты дают надежду, что скоро препарат начнут исследовании на людях и, вероятно, смогут выпустить на рынок.
Диагностика синдрома Марфана
Диагностика генетической аномалии включает в себя комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7–18 лет — это измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку в 10 см.
Генетическое обследование включает в себя выявление генотипа ДНК — идентификацию мутаций в гене FBN1. При возможности назначают специфические лабораторные тесты — определение выведения с мочой метаболитов соединительной ткани, таких как оксипролин и гликозаминогликаны.






Чтобы подтвердить нарушения развития соединительной ткани и оценить степень выраженности мутации гена FBN1, пациентам с подозрением на синдром Марфана назначают:
- ЭКГ;
- УЗИ сердца;
- КТ-ангиографию аорты и других сосудов;
- КТ грудной и брюшной полостей;
- МРТ позвоночника и головного мозга;
- специфические обследования на осмотре у офтальмолога;
- биопсию кожи.
Для окончательного определения диагноза используют общепринятые Гентские критерии 2010 года, согласно которым диагноз устанавливают в случаях:
- подтвержденной мутации гена FBN1 и расширения корня аорты или эктопией хрусталика;
- подтвержденного расширения корня аорты в сочетании с эктопией хрусталика;
- подтвержденной эктопии хрусталика в сочетании с любыми признаками системного поражения соединительной ткани.
Важно! Существует группа «марфаноподобных» синдромов, при которых внешне пациенты очень напоминают больных с аномалией гена FBN1, но причина их патологии скрывается в других нарушениях. К примеру, гомоцистинурия — это обменное заболевание, которое проявляется системными изменениями соединительной ткани, но может приводить к внезапным инсультам и существенно замедляет умственное развитие ребенка
Поэтому важно точно определить причину заболевания соединительной ткани и своевременно начать лечение.
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка
При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
С синдромом Марфана связаны не только многочисленные поводы для обращения к врачам. Часто люди с мутацией гена FBN1 компенсируют физические проявления болезни интеллектуальными способностями, поэтому это генетическое заболевание даже называют «синдромом гениев». Считается, что повышенный выброс адреналина из-за патологических изменений в надпочечниках определяет высокий тонус умственной и психической активности у таких пациентов. Именно поэтому в числе людей с синдромом Марфана можно найти известных личностей. Например, Юлию Цезарю, Аврааму Линкольну и Шарлю де Голлю патология не помешала стать известными политическими деятелями; Ганс Христиан Андерсен и Корней Чуковский создали уникальные литературные произведения, а Никколо Паганини прославился как гениальный музыкант.
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).